From IEEE Spectrum:
The Desperate Quest for Genomic Compression Algorithms
Have you had your genome sequenced yet? Millions of people around the world already have, and by 2025 that number could reach a billion.
The more genomics data that researchers acquire, the better the prospects for personal and public health. Already, prenatal DNA tests screen for developmental abnormalities. Soon, patients will have their blood sequenced to spot any nonhuman DNA that might signal an infectious disease. In the future, someone dealing with cancer will be able to track the progression of the disease by having the DNA and RNA of single cells from multiple tissues sequenced daily.
And DNA sequencing of entire populations will give us a more complete picture of society-wide health. That’s the ambition of the United Kingdom’s Biobank, which aims to sequence the genomes of 500,000 volunteers and follow them for decades. Already, population-wide genome studies are routinely used to identify mutations that correlate with specific diseases. And regular sequencing of organisms in the air, soil, and water will help track epidemics, food pathogens, toxins, and much more.
This vision will require an almost unimaginable amount of data to be stored and analyzed. Typically, a DNA sequencing machine that’s processing the entire genome of a human will generate tens to hundreds of gigabytes of data. When stored, the cumulative data of millions of genomes will occupy dozens of exabytes.
And that’s just the beginning. Scientists, physicians, and others who find genomic data useful aren’t going to stop at sequencing each individual just once [PDF]—in the same individual, they’ll want to sequence multiple cells in multiple tissues repeatedly over time. They’ll also want to sequence the DNA of other animals, plants, microorganisms, and entire ecosystems as the speed of sequencing increases and its cost falls—it’s just US $1,000 per human genome now and rapidly dropping. And the emergence of new applications—and even new industries—will compel even more sequencing.
While it’s hard to anticipate all the future benefits of genomic data, we can already see one unavoidable challenge: the nearly inconceivable amount of digital storage involved. At present the cost of storing genomic data is still just a small part of a lab’s overall budget. But that cost is growing dramatically, far outpacing the decline in the price of storage hardware. Within the next five years, the cost of storing the genomes of billions of humans, animals, plants, and microorganisms will easily hit billions of dollars per year. And this data will need to be retained for decades, if not longer.
Compressing the data obviously helps. Bioinformatics experts already use standard compression tools like gzip to shrink the size of a file by up to a factor of 20. Some researchers also use more specialized compression tools that are optimized for genomic data, but none of these tools have seen wide adoption. The two of us do research on data compression algorithms, and we think it’s time to come up with a new compression scheme—one that’s vastly more efficient, faster, and better tailored to work with the unique characteristics of genomic data. Just as special-purpose video and audio compression is essential to streaming services like YouTube and Netflix, so will targeted genomic data compression be necessary to reap the benefits of the genomic data explosion.
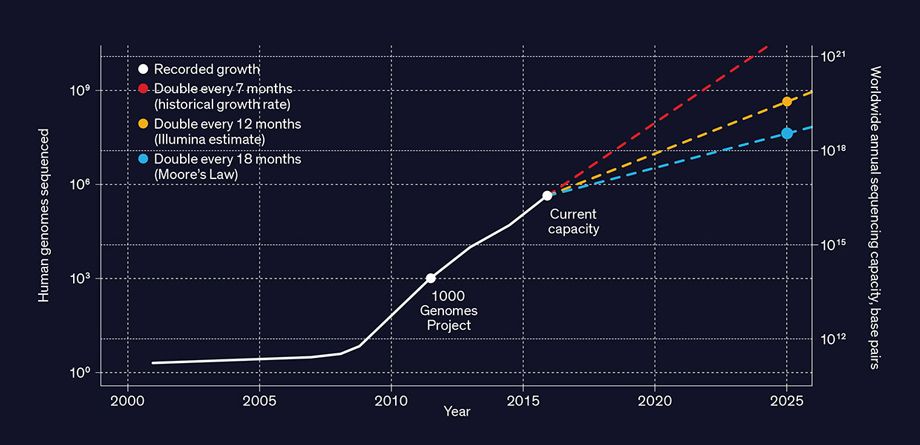
Growth of Human Genome Sequencing: Since the first
publication of a draft human genome sequence in 2001, there’s been a
dramatic increase in the pace of growth of both the number of genomes
sequenced and the sequencing capacity.
The numbers after 2015 represent
three possible projected growth curves.
Before we explain how genomic data could be better compressed, let’s take a closer look at the data itself. “Genome” here refers to the sequence of four base nucleotides—adenine, cytosine, guanine, and thymine—that compose the familiar A, C, G, T alphabet of DNA. These nucleotides occur in the chains of A-T and C-G pairs that make up the 23 pairs of chromosomes in a human genome. These chromosomes encompass some 6 billion nucleotides in most human cells and include coding genes, noncoding elements (such as the telomeres at the ends of chromosomes), regulatory elements, and mitochondrial DNA. DNA sequencing machines like those from Illumina, Oxford Nanopore Technologies, and Pacific Biosciences are able to automatically sequence a human genome from a DNA sample in hours.
These commercial DNA sequencers don’t produce a single genome-long string of ACGTs but rather a large collection of substrings, or “reads.” The reads partially overlap each other, requiring sequence-assembly software to reconstruct the full genome from them. Typically, when whole-genome sequencing is performed, each piece of the genome appears in no more than about 100 reads.
Depending on the sequencing technology used, a read can vary in length from about 100 to 100,000 base pairs, and the total number of reads varies from millions to tens of billions. Short reads can turn up single base-pair mutations, while longer reads are better for detecting complicated variations like deletions or insertions of thousands of base pairs....MORE
DNA sequencing is a noisy process, and it’s common for reads to contain errors. And so, besides the string of ACGT nucleotides, each read includes a quality score indicating the sequencing machine’s confidence in each DNA nucleotide. Sequencers express their quality scores as logarithms of error probabilities. The algorithms they use to do so are proprietary but can be checked after the fact. If a quality score is 20—corresponding to an error probability of 1 percent—a user can confirm that about 1 percent of the base pairs were incorrect in a known DNA sequence. Programs that use these files rely on quality scores to distinguish a sequencing error from, say, a mutation. A true mutation would show a higher average quality score—that is, a lower probability of error—than a sequencing error would.
The sequencer pastes together the strings and the quality scores, along with some other metadata, read by read, to form what is called a FASTQ file. A FASTQ file for an entire genome typically contains dozens to hundreds of gigabytes.
The files are also very redundant, which stems from the fact that any two human genomes are nearly identical. On average, they differ in about one nucleotide per 1,000, and it’s typically these genetic differences that are of interest. Some DNA sequencing targets specific areas of difference—for example, DNA-genotyping applications like 23andMe look only for specific variations, while DNA profiling in criminal investigations looks for variations in the number of repetitions of certain markers.
But you need to sequence the whole genome if you don’t know where the interesting stuff lies—when you’re trying to diagnose a disease of unknown genetic origin, say—and that means acquiring much larger quantities of sequencing data.
The repetition in sequencing data also comes from reading the same portions of the genome multiple times to weed out errors. Sometimes a single sample contains multiple variations of a sequence, so you’ll want to sequence it repeatedly to catch those variations. Let’s say you’re trying to detect a few cancer cells in a tissue sample or traces of fetal DNA in a pregnant woman’s blood. That may mean sequencing each DNA base pair many times, often more than 100, to distinguish the rare variations from the more common ones and also the real differences from the sequencing errors.
By now, you should have a better appreciation of why DNA sequencing generates so much redundant data. This redundancy, it turns out, is ideal for data compression. Rather than storing multiple copies of the same chunk of genomic data, you can store just one copy.
To compress genomic data, you could first divide each DNA sequence read into smaller chunks, and then assign each chunk a numerical index. Eventually, the sum total of indexes constitutes a dictionary, in which each entry isn’t a word but a short sequence of DNA base pairs.
Text compressors work this way. For example, GitHub hosts a widely used list of words that people can use to assign each word its own numerical index. So to encode a passage of text into binary, you’d replace each word with its numerical index—the list on GitHub assigns the number 64,872 to the word compression—which you’d then render in binary format. To compress the binary representation, you could sort the dictionary by word usage frequency instead of alphabetical order, so that more common words get smaller numbers and therefore take fewer bits to encode.
Another common strategy— the Lempel-Ziv family of algorithms—builds up a dictionary of progressively longer phrases rather than single words. For example, if your text often contains the word genomic followed by data, a single numerical index would be assigned to the phrase genomic data.
Many general-purpose compression tools such as gzip, bzip2, Facebook’s Zstandard, and Google’s Brotli use both of these approaches. But while these tools are good for compressing generic text, special-purpose compressors built to exploit patterns in certain kinds of data can dramatically outperform them.
Consider the case of streaming video. A single frame of a video and the direction of its motion enable video compression software to predict the next frame, so the compressed file won’t include the data for every pixel of every frame....